


¿Conoces la enfermedad de Wilson?
- Escrito por Dra. Fernanda García Alvarado
- Publicado en Hígado Graso Metabólico
La enfermedad de Wilson es una enfermedad rara que puede manifestarse como enfermedad hepática, neurológica o psiquiátrica. Se hereda a la vez del padre y de la madre (herencia autonómica recesiva) por mutaciones en un gen ATP7B, localizado en el cromosoma 13, que codifica una proteína necesaria para eliminar el cobre sobrante desde el interior de la célula hepática a la bilis.
Esto provoca la acumulación tóxica de cobre procedente de la dieta en el organismo. Afecta, sobre todo, a hígado y cerebro. La enfermedad de Wilson afecta entre 10 y 30 casos por cada millón de habitantes. Los síntomas aparecen en la mayoría de los pacientes entre los cinco y los 40 años.
En condiciones normales la mayor parte del cobre ingerido (2-5 miligramos/día) se elimina por la bilis y sólo una pequeña cantidad por la orina. Las recomendaciones específicas dependen de la edad, el sexo y factores como el embarazo.
El cobre es un oligoelemento esencial presente en todos los tejidos del cuerpo y que trabaja en la formación de los glóbulos rojos además de favorecer la buena salud de los vasos sanguíneos, los nervios, el sistema inmunitario y los huesos. También ayuda a la absorción del hierro.
Los afectados de enfermedad de Wilson tienen una disminución de la excreción biliar del cobre, por una mutación genética lo que produce la acumulación tóxica de este micronutrimento en el organismo que puede dañar hígado, cerebro, riñones e, incluso, los ojos. Se han descrito más de 300 mutaciones del gen ATP7B implicadas en el desarrollo de la enfermedad de Wilson.
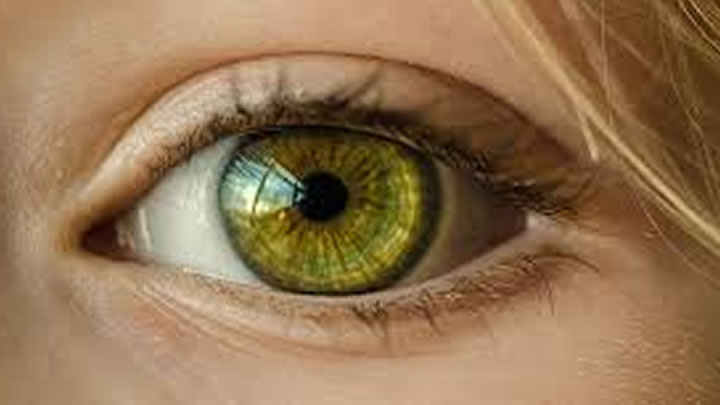
El diagnóstico clínico se basa en el examen físico, la descripción de síntomas y, el análisis ocular de la existencia o no del anillo de Kayser-Fleischer, una franja oscura de color de dorado a verdoso situada en la periferia de la córnea.
El tratamiento posterior consiste en evitar que el cobre vuelva a acumularse. El tratamiento de referencia son los medicamentos quelantes, que fijan el cobre y facilitan su eliminación. Cuando el daño hepático es grave, podría ser necesario realizar un trasplante de hígado.
En México, se considera un padecimiento raro o poco frecuente. Por ello, en caso de tener un padecimiento crónico del hígado que no sea por alcohol, ni viral, ni autoinmune, ni ligado a enfermedad por hígado graso, debes acudir a un médico especialista en enfermedades del hígado, para que considere realizar las pruebas de esta enfermedad. En general se requiere disponer de al menos una determinación de cobre en suero, en orina y la cuantificación de la proteína transportadora del cobre (la ceruloplasmiana). En ocasiones será necesario realizar una biopsia, lo cual permite identificar al cobre, gracias a tinciones especiales.
 En Costa Rica se han descrito una gran cantidad casos, en población pediátrica. En una publicación del Dr. Jiménez y colaboradores, del Hospital Nacional de Niños, "Doctor Carlos Saenz Herrera" asi como del Children's Hospital, de la Universidad de Ontario, Canadá, se describieron 35 niños, de los cuales un 69% eran varones. La edad promedio mento de presentación era de 8 a 12 años, (rango, 5–15).
En Costa Rica se han descrito una gran cantidad casos, en población pediátrica. En una publicación del Dr. Jiménez y colaboradores, del Hospital Nacional de Niños, "Doctor Carlos Saenz Herrera" asi como del Children's Hospital, de la Universidad de Ontario, Canadá, se describieron 35 niños, de los cuales un 69% eran varones. La edad promedio mento de presentación era de 8 a 12 años, (rango, 5–15).
En la presentación clínica se incluían síntomas hepáticos en el 69% de los casos, hematológicos en el11% y neurológicos en el 3%. Seis (17%) eran hermanos asintomáticos.Todos tuvieron valores bajos de ceruloplasmina sérica. Los valores de las transaminasas resultaron anormales en el 50% de los pacientes. Los valores se ́ricos de albúmina y TP fueron normales en todos ellos. La excreción de cobre en orina de 24 h estuvo elevada en todos los pacientes. Ninguno de los niños presentó anillo de K-F. Se realizaron biopsias hepáticas en 21 pacientes (60%), que revelaron la presencia de cirrosis en 5 de ellos. Se llevó a cabo un trasplante hepático en 5 pacientes debido a una fallo hepático agudo. Seis niños (17%) fallecieron a causa de fallo hepático fulminante incontrolable.
Los autores concluyen que la Enfermedad de Wilson debe sospecharse en niños con anormalidades persistentes de la función hepática.
Referencias:
1. Członkowska, A., Litwin, T., Dusek, P., Ferenci, P., Lutsenko, S., Medici, V., ... & Schilsky, M. L. Wilson disease. Nature Reviews Disease Primers, 2018:4(1), 21.
2. Jimenez G, Cambronero V, Morales C, Mora A, Guzman C, Jimenez-Riverab C. Enfermedad de Wilson: experiencia pediatrica en Costa Rica. Gastroenterol Hepatol. 2009;32(4):274–278.
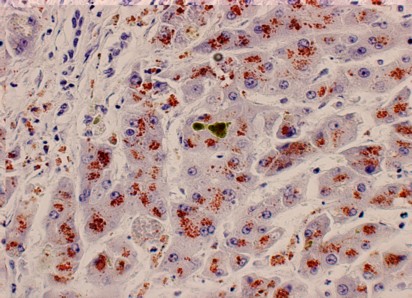
3. Rashmil Saxena. Special Stains in Interpretation of Liver Biopsies. Technical Articles. Connection 2010;92-103. Imagen microscópica del hígado, con tinción de Orceina, de un paciente con Enfermedad de Wilson. Se observan gránulos rojizos en el interior de las células que corresponde a exceso de cobre. Es una publicación del Departamento de Cirugía, de la Universidad de Indiana, Indianapolis, USA.
Artículo de Divulgación revisado y adaptado por el Dr. Jorge Luis Poo, Hepatólogo Clínico, miembro del Comité Editorial de tu portal AMHIGO y fundador del Grupo Mexicano para el Estudio de las Enfermedades Hepáticas